Ideas and Results
Combinatorics
Originally we looked into the combinatorial aspect of the problem, hoping for some usable results regarding the number of potentially functional siRNA, based on favorable properties such as G/C content, low level of secondary structure, differential in A/U content between 5' and 3' ends, and the absence of long G/C stretches in the strings.
Taking these conditions into consideration narrowed down the total number of possible siRNA strings of a certain length (in our case, of length n=20) to 25% of the original population. Particularly, we counted strings of siRNA of length 20 which satisfied the following conditions:
- G/C content between six and eleven nts
- A/U content at 3' end greater than A/U content at 5' end, where we considered four nts at each end
- No consecutive G/C stretches of six nts or longer
The formula for determining the number of potentially functional sequences using this method is given below:
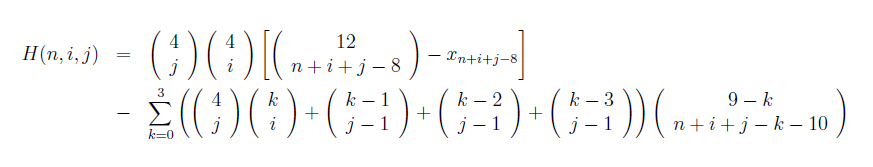
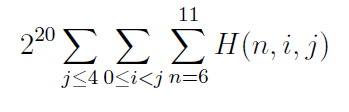
While we were able to narrow down the potential siRNA strings to approximately 25% of the total number of possible strings, this was still a huge number--somewhere on the order of 3 x 10 11. We considered taking the secondary structure of siRNA into consideration--that is, to consider the two-dimensional shape of the strings in addition to the nucleotides themselves, but this method requires knowing the secondary structure of the binding sites on the mRNA for any given binding site, and so we decided to try a different tact.
Levenshtein Distance Again
Towards the beginning of the program we ran some test strings through an unweighted Levenshtein Distance calculator (web-based implementations are easy to find) with relatively poor results, so it seemed that there are more factors than substitutions/mutations to consider. For example, the position of the substitution seemed to be quite important, given the "ideal conditions" for functionality presented above.
Vert et al's 2006 paper includes a table which specifies weights for each nucleotide at each position of a 21-nt string based on functionality, which you can see here. Using these weights, we modified the Levenshtein Distance so that it would reflect the change in functionality of a substitution of each nucleotide for another at every position and implemented it as a program in C++.
To test the accuracy of the algorithm, we developed a program (HEK.exe), and a general description of the program and how it works can be found in the walkthrough. We have not yet finished running all of our data sets through the program yet, but preliminary results are promising.